

Kutane Sarkome
Zuletzt aktualisiert: 25.06.2024 | Autor(in): Prof. Dr. med. Friedegund Meier
Kutane Sarkome entstehen aus bösartigen mesenchymalen Zellen, d.h. Stammzellen des Binde- und Stützgewebes in der Leder- oder/und Unterhaut. Kutane Sarkome sind deutlich seltener als der helle oder schwarze Hautkrebs . Im Vergleich zu Sarkomen der tieferliegenden Binde- und Stützgewebe haben kutane Sarkome meistens eine günstigere Prognose, sie bilden seltener Metastasen (Absiedelungen). Zu den häufigsten kutanen Sarkomen zählen das atypische Fibroxanthom (AFX), das pleomorphe dermale Sarkom (PDS), das Dermatofibrosarcoma protuberans (DFSP), das kutane Angiosarkom, das Kaposi-Sarkom und das kutane Leiomyosarkom. Hierfür finden Sie leitlinienbasierte Informationen im Infoportal Hautkrebs. Da kutane Sarkome seltener sind, gibt es nicht für alle kutanen Sarkome Leitlinien mit Standards für Diagnostik, Therapie und Nachsorge.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Dermales und subkutanes Leiomyosarkom
Zuletzt aktualisiert: 24.06.2026 | Autor(in): Doris Helbig
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Dermales und subkutanes Leiomyosarkom
Zuletzt aktualisiert: 24.06.2026 | Autor(in): Doris Helbig
Superfizielle Leiomyosarkome (LMS) sind seltene Tumoren. Diese gehen von Strukturen der Unterhaut (dermales/kutanes Leiomyosarkom) oder des Unterhautfettgewebes (subkutanes LMS) aus (2-3% aller Sarkome) (1-3). Charakteristischerweise sind Personen im Alter von 50-80 Jahren (subkutane LMS tendenziell bei älteren Personen) betroffen (4-8).
Darüber hinaus können primäre superfizielle LMS sehr selten im Rahmen syndromaler Krankheitsbilder auftreten (Reed Syndrom, familiäre/hereditäre Leiomyomatose, Birt-Hogg-Dube Syndrom sowie Li-Fraumeni Syndrom).
Dermale und subkutane Leiomyosarkome (LMS) sind seltene bösartige Tumoren der Haut.
Sie unterscheiden sich im Wesentlichen durch ihre Entstehung in unterschiedlich tiefen Hautschichten.
- REFERENZEN
- Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S1-Leitlinie Dermales und subkutanes Leiomyosarkom (LMS)
- Aneiros-Fernandez J, Antonio Retamero J, Husein-Elahmed H, Ovalle F, Aneiros-Cachaza J. Primary cutaneous and subcutaneous leiomyosarcomas: evolution and prognostic factors. Eur J Dermatol. 2016;26(1):9-12.
- Rouhani P, Fletcher CD, Devesa SS, Toro JR. Cutaneous soft tissue sarcoma incidence patterns in the U.S. : an analysis of 12,114 cases. Cancer. 2008;113(3):616-27.
- Kazlouskaya V, Lai YC, Khachemoune A. Leiomyosarcoma of the skin: review of the literature with an emphasis on prognosis and management. Int J Dermatol. 2020;59(2):165-72.
- Holst VA, Junkins-Hopkins JM, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46(4):477-90; quiz, 91-4.
- Winchester DS, Hocker TL, Brewer JD, Baum CL, Hochwalt PC, Arpey CJ, et al. Leiomyosarcoma of the skin: clinical, histopathologic, and prognostic factors that influence outcomes. J Am Acad Dermatol. 2014;71(5):919-25.
- Murback NDN, Takita LC, Castro BC, Filho GH. Cutaneous leiomyosarcoma on the face. An Bras Dermatol. 2018;93(2):262-4.
- Ortins-Pina A, Soares-de-Almeida L, Rutten A. Primary cutaneous vascular leiomyosarcoma: A rare subtype of leiomyosarcoma of the skin. J Cutan Pathol. 2018;45(8):639-41.
- Deneve JL, Messina JL, Bui MM, Marzban SS, Letson GD, Cheong D, et al. Cutaneous leiomyosarcoma: treatment and outcomes with a standardized margin of resection. Cancer Control. 2013;20(4):307-12.
- Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73(1):95-106.
- Bird LM, Kuo DJ, Masser-Frye D, Mo JQ, Elster JD. Leiomyosarcoma in Birt-Hogg-Dube Syndrome. J Pediatr Hematol Oncol. 2020;42(2):136-7.
- Sabater-Marco V, Ferrando-Roca F, Morera-Faet A, Garcia-Garcia JA, Bosch SB, Lopez-Guerrero JA. Primary Cutaneous Leiomyosarcoma Arising in a Patient With Li-Fraumeni Syndrome: A Neoplasm With Unusual Histopathologic Features and Loss of Heterozygosity at TP53 Gene. Am J Dermatopathol. 2018;40(3):225-7.
- Fauth CT, Bruecks AK, Temple W, Arlette JP, DiFrancesco LM. Superficial leiomyosarcoma: a clinicopathologic review and update. J Cutan Pathol. 2010;37(2):269-76.
- Kraft S, Fletcher CD. Atypical intradermal smooth muscle neoplasms: clinicopathologic analysis of 84 cases and a reappraisal of cutaneous "leiomyosarcoma". Am J Surg Pathol. 2011;35(4):599-607.
- Wong GN, Webb A, Gyorki D, McCormack C, Tran P, Ngan SY, et al. Cutaneous leiomyosarcoma: dermal and subcutaneous. Australas J Dermatol. 2020;61(3):243-9.
- Massi D, Franchi A, Alos L, Cook M, Di Palma S, Enguita AB, et al. Primary cutaneous leiomyosarcoma: clinicopathological analysis of 36 cases. Histopathology. 2010;56(2):251-62.
- Kohlmeyer J, Steimle-Grauer SA, Hein R. Cutaneous sarcomas. J Dtsch Dermatol Ges. 2017;15(6):630-48.
- Fields JP, Helwig EB. Leiomyosarcoma of the skin and subcutaneous tissue. Cancer. 1981;47(1):156-69.
- Hollmig ST, Sachdev R, Cockerell CJ, Posten W, Chiang M, Kim J. Spindle cell neoplasms encountered in dermatologic surgery: a review. Dermatol Surg. 2012;38(6):825-50.
- Zacher M, Heppt MV, Brinker TJ, Hayani KM, Flaig MJ, Berking C. Primary leiomyosarcoma of the skin: a comprehensive review on diagnosis and treatment. Med Oncol. 2018;35(10):135.
- Wascher RA, Lee MY. Recurrent cutaneous leiomyosarcoma. Cancer. 1992;70(2):490-2.
- Svarvar C, Bohling T, Berlin O, Gustafson P, Folleras G, Bjerkehagen B, et al. Clinical course of nonvisceral soft tissue leiomyosarcoma in 225 patients from the Scandinavian Sarcoma Group. Cancer. 2007;109(2):282-91.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Klinik und Prognose
Zuletzt aktualisiert: 24.06.2026 | Autor(in): PD Dr. med. Doris Helbig
LMS treten typischerweise an den Beinen, am Rumpf oder am Kopf auf. Sie präsentieren sich als schmerzhafte, rötliche bis bräunliche Knoten (4, 12-14).
Hinsichtlich Prognose und Behandlung unterscheiden sich dermale (oberflächiger, in der Unterhaut) und subkutane LMS (tiefer, im Unterhautfettgewebe). Nach vollständiger chirurgischer Entfernung eines dermalen LMS mit einem Mindestsicherheitsabstand von 1 cm treten Rezidive (=Rückfälle) am Ort des Primärtumors oder Metastasen sehr selten auf (in der Regel 0%). Bei subkutanen LMS sind Rezidive bzw. Metastasen möglich, letztere streuen am häufigsten in weiter entfernte Hautareale oder die Lunge. Deshalb sollte hier ein ausreichender chirurgischer Sicherheitsabstand oder ein interdisziplinäres Vorgehen (ggf. Nachbestrahlung) gewählt werden (1, 5, 15, 16).
Entsprechend hat die Nachsorge von Patient:innen mit LMS vor allem die frühzeitige Erfassung von Lokalrezidiven, Hautmetastasen oder Fernmetastasen zum Ziel. Bei einem dermalen LMS erscheinen halbjährliche klinische Untersuchungen einschließlich der Abtastung der Lymphknoten der Region, anschließend jährlich für mindestens fünf Jahre empfehlenswert. Bei subkutanen LMS sollten klinische Untersuchungen in Abständen von 3 Monaten innerhalb der ersten beiden Jahre und anschließend alle 6 Monate für mindestens fünf Jahre erfolgen. Zusätzlich wird eine Ultraschalluntersuchung der ursprünglichen Tumorregion sowie der lokalen Lymphknoten in 6-monatlichen Abständen empfohlen. Weiterführende apparative Untersuchungen wie Computertomografie (CT) erscheinen lediglich bei Auffälligkeiten in der Sonografie oder bei subkutanen LMS mit Risikofaktoren, Rezidiven oder bereits metastasierten Tumoren angezeigt.
LMS treten typischerweise an den Beinen, am Rumpf oder Kopf auf. Sie präsentieren sich als schmerzhafte, rötliche bis bräunliche Knoten.
Nach vollständiger Entfernung des Tumors mit einem Mindestsicherheitsabstand von 1 cm treten Rückfälle bzw. Metastasen bei dermalen LMS sehr selten auf. Bei subkutanen LMS sind Rezidive bzw. Metastasen möglich, letztere am häufigsten in weiter entfernte Hautareale oder die Lunge.
Die Nachsorge erfolgt in regelmäßigen Abständen. Sie ist vor allem auf den Nachweis eines lokalen Rückfalls ausgerichtet.
- REFERENZEN
- Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S1-Leitlinie Dermales und subkutanes Leiomyosarkom (LMS)
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Therapie
Zuletzt aktualisiert: 24.06.2026 | Autor(in): PD Dr. med. Doris Helbig
Für das kutane und subkutane LMS soll unter kurativer (=heilender) Absicht eine radikale Exzision mit anschließender feingeweblicher Aufarbeitung erfolgen. Wenn möglich soll eine sogenannte mikrographisch kontrollierte Exzision (MKC) erfolgen, sowie ein entsprechender Sicherheitsabstand zum Tumorrand eingehalten werden. Beim dermalen LMS erscheint ein Sicherheitsabstand von 1 cm ausreichend. Beim subkutanen LMS sollte der Sicherheitsabstand ggf. erweitert werden (unter Berücksichtigung anatomischer, funktioneller und ästhetischer Aspekte), um das lokale Rezidivrisiko zu senken. Die endgültige Entscheidung sollte der/die Operateur:in nach interdisziplinärer Tumorkonferenz im Einverständnis mit dem/der informierten Patient:in treffen (3-5, 8, 17).
Bei superfiziellen LMS im Rahmen syndromaler Krankheitsbilder (Reed Syndrom, familiäre/hereditäre Leiomyomatose, Birt-Hogg-Dube Syndrom sowie Li-Fraumeni Syndrom) sollten die Eigen- und Familienkrebsanamnese erhoben sowie ggf. weitere Malignome mittels Schnittbilddiagnostik ausgeschlossen werden.
Die Entscheidung einer Strahlentherapie sollte nach sorgfältiger Abwägung im Rahmen einer interdisziplinären Tumorboardbesprechung getroffen werden. Eine Bestrahlung ist bei kleinen, vollständig entfernten Tumoren nicht angezeigt. Die Bestrahlung wird aber bei nicht vollständig entfernten oder großen LMS (> 5cm) in Betracht gezogen (18, 19). Auch wenn der Sicherheitsabstand zum Tumorrand gering ist, kann eine zusätzliche (=adjuvante) Bestrahlung erwogen werden. Bei subkutanen LMS (16, 18, 20) sowie nach Rezidivresektion kann ein Mehrwert der Bestrahlung angenommenwerden, lässt sich aber anhand der vorliegenden Daten nicht beweisen. Zur definitiven oder neoadjuvanten Bestrahlung bei primärer Inoperabilität liegen bislang keine Daten vor. Eine Übertragbarkeit der Daten von anderen Sarkomen kann nur vermutet werden.
Medikamentöse Therapie
Es liegen wenig Daten zur Systemtherapie (=medikamentöse Therapie) superfizieller LMS vor. Therapieempfehlungen für inoperable oder metastasierte Patient:innen sollten im Rahmen eines interdisziplinären Tumorboards individuell besprochen werden. In die Entscheidung könnten neben den klinischen Aspekten, Ergebnisse molekulargenetischer Untersuchungen und mikroskopische Parameter einfließen. Es gibt vereinzelte Berichte über Behandlungsversuche mit verschiedenen Chemotherapien (14, 21).
Die Therapie der ersten Wahl ist die vollständige, jedoch möglichst funktionserhaltende chirurgische Entfernung. Sie sollte mit Sicherheitsabstand und mikrographisch kontrollierter Chirurgie (MKC) erfolgen.
Die Entscheidung zu einer Strahlentherapie sollte nach sorgfältiger Abwägung im Rahmen einer interdisziplinären Tumorboardbesprechung getroffen werden.
Eine wirksame medikamentöse Standardtherapie fortgeschrittener LMS ist nicht bekannt.
- REFERENZEN
- Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S1-Leitlinie Dermales und subkutanes Leiomyosarkom (LMS)
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Kutane Angiosarkome
Zuletzt aktualisiert: 10.02.2025 | Autor(in): Thomas Vogt
Kutane Angiosarkome sind seltene, bösartige Tumoren der Blut- und Lymphgefäße der Haut. Sie treten bei älteren Menschen auf, in der Regel sind die Betroffenen über 70 Jahre alt. Angiosarkome entstehen meist an der Kopfhaut und im Gesicht und zeigen sich zunächst als einfache rot-blaue Flecken.
Was ist ein Angiosarkom?
Angiosarkome sind seltene (0,4 Fälle pro 1 Mio. EW / Jahr), bösartige Tumoren der Blut- bzw. Lymphgefäße der Haut. Sie kommen vornehmlich sporadisch, ohne fassbare Ursache vor, sie können aber auch einige Jahre nach einer Strahlentherapie (z.B. an der Brust nach Mammakarzinom) im Strahlenfeld vorkommen. Die Tumoren treten überwiegend an der Kopfhaut und im Gesicht in Erscheinung, zunächst als „unspektakuläre“ rötlich-bläuliche Verfärbungen, also Flecke, die zu Beginn der Krebserkrankung regelmäßig verkannt werden als banale Entzündungen oder Blutergüsse. Erst sehr spät bildet das Sarkom Knoten, die blutend aufbrechen und dann nicht selten schon Metastasen, z.B. in der Lunge, verursacht haben. Männer sind zweimal häufiger betroffen als Frauen und die Betroffenen sind in der Regel über 70 Jahre alt.
Wie wird ein Angiosarkom diagnostiziert?
Zunächst muss eine Ärztin oder ein Arzt überhaupt bei „unklaren Rötungen“ an diese seltene, aber gefährliche Tumorerkrankung denken. Die Diagnose stellt man anhand einer oder mehrerer kleiner Biopsien. In den Proben können die blitzförmigen, das Gewebe diffus durchsetzenden, bösartigen Gefäßstrukturen nachgewiesen werden, und mit einigen Spezialfärbungen kann die Diagnose mit sehr hoher Treffsicherheit schnell gestellt werden.
Zur Ausbreitungsdiagnostik muss vor allem eine genaue klinische Untersuchung der Haut, ggf. mit zusätzlichen Biopsien zur Ausdehnungsbestimmung, erfolgen. Zum Metastasenausschluss sind alle gängigen bildgebenden Verfahren wie Kernspinuntersuchung oder CT und auch das sog. PET-CT geeignet.
Wie ist die Prognose bei einem Angiosarkom?
Auch hier gilt wie bei vielen Tumoren, es ist eine Frage der Größe des Tumors und der Ausdehnung der Erkrankung zum Diagnosezeitpunkt. Kleinere (< 5 cm), früh diagnostizierte Tumoren, die komplett operiert und nachbestrahlt werden können, haben entgegen vieler Behauptungen keine schlechte Prognose. Die Prognose bei flächig ausgedehnteren Tumoren, die nicht vollständig entfernt und gut nachbestrahlt werden können, ist allerdings leider immer noch sehr ernst. So werden für lokal ausgedehnte Angiosarkome 3-Jahresüberlebensraten um 40% und 10-Jahresraten um 20% angegeben.
Operative Behandlung des Angiosarkoms
Bei nicht zu großer Ausdehnung wird heute an erster Stelle eine Exzision (Entferung) möglichst ohne Zurücklassung von versprengtem Resttumorgewebe angestrebt. In der Praxis sind hier allerdings, man denke z.B. an eine Ausdehnung über das halbe Gesicht, Grenzen gesetzt und zusätzlich streut der Tumor oft kleine sogenannte „skip“-Läsionen in die direkte Umgebung, so dass die Gefahr von Rückfällen bei alleiniger sehr hoch ist. Dementsprechend werden Angiosarkom-OP Gebiete nach der Wundheilung möglichst zusätzlich vorbeugend nachbestrahlt.
Nicht-operative Behandlung des Angiosarkoms
Bei zu ausgedehnten Tumoren, die nicht komplett operiert werden können oder bei Rückfällen bzw. metastatischen Verläufen müssen im interdisziplinären Tumorboard folgende Behandlungsmöglichkeiten allein, in Kombination oder nacheinander abgewogen werden:
- Tumor - verkleinernde Operationen,
- die primäre alleinige Bestrahlung,
- die sog. Elektrochemotherapie,
- die Radiochemotherapie
- oder auch primäre rein medikamentöse Therapien unter Einbeziehung der Immuncheckpointinhibition (siehe Immuntherapie).
Die führende medikamentöse Therapie ist aktuell in erster Linie die Gabe des recht gut tolerierten Chemotherapeutikums Paclitaxel. Die Tumoren sprechen gut an, es kommt bei Absetzen aber oft schnell zu Rückfällen. Daher wird heute auch eine langzeitig fortgesetzte Chemotherapie („maintenance chemotherapy“) in niedrigen Dosierungen und längeren Intervallen, ggf. unterbrochen durch „drug holidays“, diskutiert, d.h. eine Therapie bis die Erkrankung wiederkommt, fortschreitet oder Nebenwirkungen der Therapie zum Abbruch zwingen.
Weitere Behandlungsmöglichkeiten des Angiosarkoms
Es gibt neue molekular gezielte Therapien, wenn die erste Chemotherapie versagt: In dieser sog. „second line“ ist bspw. Pazopanib, ein Signalübertragungshemmer in Tablettenform, zugelassen und ebenfalls das Trabectedin, eine Substanz aus Tiefseetunikaten, welche die Tumorzell-DNA schädigt.
Hoffnung auf noch bessere Therapieerfolge machen weltweit aktuelle Studien, die vorrangig das o.g. Paclitaxel mit einer Immuntherapie kombinieren, genauer gesagt mit der Immuncheckpoint-Inhibitor-Therapie, speziell sog. Anti-PD(L)1 Antikörpern. Letztere Therapeutika nutzen das körpereigene Abwehrsystem, um die Tumorzellen zu vernichten. Diese durch einen Medizinnobelpreis ausgezeichnete Therapiestrategie verbessert heute die Überlebenschancen vieler Krebspatienten mit ganz unterschiedlichen Krebsarten und auch die der Angiosarkompatienten.
Das Angiosarkom ist ein seltener, anfangs unscheinbarer, oft als einfacher rot - blauer Fleck erscheinender, aber sehr gefährlicher Tumor der Haut vor allem bei älteren Männern am Kopf (Gesicht und Kopfhaut).
Bei kleineren Tumoren (<5cm Durchmesser), die frühzeitig erkannt werden, kann , wenn sie komplett operiert und nachbestrahlt werden können, ein langfristiges Überleben der Erkrankung möglich sein.
Ausgedehnte oder gar metastasierte Angiosarkome können ebenfalls mit diversen modernen Instrumenten heute schon recht gut behandelt werden, z.B. durch Chemotherapie, Elektrochemotherapie, molekular gezielte Therapie. Eine langfristige Heilung ist aber dann nur noch in wenigen Fällen möglich. Neue Hoffnung macht auch hier, wie bei vielen anderen Krebsarten, vor allem der Einsatz der Immuntherapie mit Antikörpern derzeit noch innerhalb klinischer Studien oder im „off label“ - Einsatz im Rahmen eines individuellen Heilversuchs, der von Ihren betreuenden Ärzten beantragt werden muss.
- REFERENZEN
- [1] Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S1-Leitlinie Kutane Angiosarkome Registernummer 032 – 056; Version: 1.0 Stand: 04.01.2021Letzter Zugriff: Gültig bis: 03.01.2026
- [2] Vogt T, Melchior P, Rübe C, Ugurel S, Schimming TT, Utikal J, Esser S, Helbig D, Hadaschik E, Kasper B, Grabbe S.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Dermatofibrosarcoma protuberans (DFSP)
Zuletzt aktualisiert: 22.04.2025 | Autor(in): Prof. Dr. med. Selma Ugurel
Urspung und Häufigkeit
Das Dermatofibrosarcoma protuberans (DFSP) ist ein bindegewebsartiger Tumor, der von der Haut ausgeht. Es handelt sich um einen Tumor von geringer Malignität (Bösartigkeit). Dies bedeutet, dass das DFSP zwar unkontrolliert wächst, und dadurch umgebende Strukturen wie Unterhaut, Muskel oder Knochen verdrängen oder zerstören kann, aber nur selten metastasiert (in die Ferne ausstreut).
Das DFSP ist ein seltener Tumor (unter 1 pro 100.000 Einwohner und Jahr sind betroffen). Das Durchschnittsalter der Patient:innen liegt bei 40 Jahren. DFSP können auch im Kindesalter vorkommen, dies ist aber extrem selten. Männer und Frauen sind gleich häufig betroffen.
Symptome und Verlauf
Das DFSP ist beim Patienten zumeist als hautfarbener bis rötlicher, manchmal auch braun-gelblich gefärbter, flach erhabener, verhärteter, unregelmäßig begrenzter Tumor sichtbar. Typischerweise wächst das DFSP sehr langsam, über Jahre bis Jahrzehnte. Im Verlauf entstehen dabei häufig einer oder mehrere Tumorknoten auf einem flächig ausgedehnten Tumorgrund. Meist befindet sich das DFSP an Brust, Rücken, Oberarmen oder Oberschenkeln.
Das DFSP neigt dazu, nach einer Operation wiederzukehren (Lokalrezidiv), streut aber nur selten aus (Metastasierungsrate \<1% der Fälle). Eine Sonderform, das fibrosarkomatös transformierte DFSP (DFSP-FS), ist eine bösartigere Form des DFSP und weist eine höhere Metastasierungsrate auf. Eine weitere Sonderform ist die bräunlich gefärbte Variante des DFSP. Sie wird auch als Bednar-Tumor bezeichnet, und ist nicht durch höhere Bösartigkeit gekennzeichnet.
Diagnostik
Eine sichere Diagnose des DFSP ist durch die körperliche Untersuchung nicht möglich. Die Diagnose muss durch eine Hautprobe (Biopsie) mit nachfolgender feingeweblicher Aufarbeitung (Histologie) gestellt werden. Der für das DFSP typische Gewebemarker in der histologischen Untersuchung ist CD34. Die Mehrzahl der DFSP tragen chromosomale Störungen, sogenannte Translokationen. Die häufigste ist die Translokation 17q22; 22q13, die mit einer Fusion der Gene COL1A1 und PDGF-beta einhergeht. Diese Chromosomenstörungen kann man mittels molekularpathologischer Verfahren nachweisen. Sie können für eine medikamentöse Behandlung bedeutsam sein. Um festzustellen, ob sich ein DFSP bereits ausgebreitet hat, kann es sinnvoll sein Untersuchungen wie Ultraschall, Computer-Tomographie, oder Magnetresonanz-Tomographie durchführen zu lassen.
Therapie mittels Operation
Die allererste empfohlene Therapie nach Entdeckung eines DFSP ist die komplette chirurgische Entfernung (Operation). Hierbei wird zusätzlich zum erkrankten Tumorgewebe auch ein Stück des umliegenden gesunden Gewebes entfernt (Sicherheitsabstand), um das Risiko eines Wiederauftretens (Lokalrezidiv) zu verringern.
Lokalrezidive sind schwierig zu erkennen, da das durch die erste Operation entstandene Narbengewebe dem Aussehen der DFSP Tumore ähnelt.
Therapie mittels Bestrahlung
Das DFSP gilt als strahlenempfindlicher Tumor. Daher ist eine Strahlentherapie eine gute Behandlungsoption für Patient:innen, deren Tumore nicht mehr oder nicht komplett operiert werden können. Auch nach Wiederauftreten (Lokalrezidiv) eines DFSP kann es sinnvoll sein, die betroffene Stelle nachzubestrahlen. Eine generelle Empfehlung für eine Nachbestrahlung komplett operierter DFSP (adjuvante Bestrahlung) gibt es jedoch nicht.
Therapie mittels Medikamenten
Wenn DFSP Tumore nicht mehr operabel sind, kann eine Behandlung mit Medikamenten notwendig werden.
Eine Chemotherapie ist beim DFSP wenig wirksam und wird daher nicht zur Erstbehandlung empfohlen.
Eine molekular zielgerichtete Therapie mit Stoffwechselblockern (Blocker von PDGF-R) kann in etwa der Hälfte der betroffenen Patient:innen eine Verkleinerung der Tumore erzielen. Das hierfür zugelassene Präparat ist Imatinib; es wird in Tablettenform eingenommen. Die PDGF-R Blocker wirken auch bei den bösartigeren fibrosarkomatös transformierten DFSP (DFSP-FS). Nach erfolgreicher medikamentöser Behandlung können die DFSP Tumore soweit verkleinert werden, dass eine Operation möglich wird.
Nachsorge
Für Patient:innen mit DFSP ist es vor allem wichtig, ein Wiederauftreten der Tumoren am Ort der vorangegangenen Operation (Lokalrezidiv) frühzeitig zu erkennen. Hierzu ist es ausreichend, eine regelmäßige Untersuchung der Haut durchzuführen, apparative Untersuchungen sind nur bei Patient:innen empfehlenswert, die bereits eine Tumorausstreuung (Metastasierung) hatten. Die betroffenen Patient:innen sollten auch selbst auf Veränderungen achten, die ein Lokalrezidiv anzeigen könnten (Schwellungen oder Verhärtungen in oder unter der Haut in der Region der vorangegangenen Operation).
Das Dermatofibrosarkom (DFSP) ist ein seltener Hautkrebs, der unspezifisch aussieht (hautfarbig bis rot) und im mittleren Lebensalter auftritt (Durchschnittsalter 40 Jahre).
Als erste Therapie wird eine komplette chirurgische Entfernung empfohlen, mit Sicherheitsabstand zum gesunden Gewebe um das Risiko eines Lokalrezidivs zu verringern.
Eine gezielte molekulare Therapie mit PDGFR Hemmern ist eine mögliche Behandlung bei nicht mehr operablen DFSP
- REFERENZEN
- [1] J Dtsch Dermatol Ges 2019 Jun;17(6):663-668. doi: 10.1111/ddg.13849_g. S1-Leitlinie Dermatofibrosarcoma protuberans (DFSP) - Update 2018 Selma Ugurel 1, Rolf-Dieter Kortmann 2, Peter Mohr 3, Thomas Mentzel 4, Claus Garbe 5, Helmut Breuninger 5, Sebastian Bauer 6, Stephan Grabbe 7
- [2] Mentzel T. Fibrohistiocytic tumors of the skin: a heterogeneous group of superficially located mesenchymal neoplasms. Pathologe. 2015;36:79-88. Shimizu A, O'Brien KP, Sjoblom T, et al. The dermatofibrosarcoma protuberans-associated collagen type Ialpha1/platelet-derived growth factor (PDGF) B-chain fusion gene generates a transforming protein that is processed to functional PDGF-BB. Cancer Res. 1999;59:3719-23. Trofymenko O, Bordeaux JS, Zeitouni NC. Survival in patients with primary dermatofibrosarcoma protuberans: National Cancer Database analysis. J Am Acad Dermatol. 2018;78:1125-34. Rutkowski P, Van GM, Rankin CJ, et al. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol. 2010;28:1772-9. Ugurel S, Mentzel T, Utikal J, et al. Neoadjuvant imatinib in advanced primary or locally recurrent dermatofibrosarcoma protuberans: a multicenter phase II DeCOG trial with long-term follow-up. Clin Cancer Res. 2014;20:499-510.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Atypisches Fibroxanthom und Pleomorphes Dermales Sarkom
Zuletzt aktualisiert: 14.01.2026 | Autor(in): Doris Helbig, Mirjana Ziemer
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Atypisches Fibroxanthom und Pleomorphes Dermales Sarkom
Zuletzt aktualisiert: 14.01.2026 | Autor(in): Doris Helbig, Mirjana Ziemer
Verbreitung und Ursachen
Das atypische Fibroxanthom (AFX) und das pleomorphe dermale Sarkom (PDS) sind Spektren einer seltenen bösartigen Erkrankung der Haut. Genaue Daten zur Häufigkeit gibt es nicht. Die Diagnose des AFX wurde erstmal in den 1960er-Jahren beschrieben.
Über den Ursprung der Tumorzellen wird seit Erstbeschreibung diskutiert. Das liegt nicht zuletzt auch daran, dass in den zurückliegenden Jahrzehnten irrtümlich andere mikroskopisch ähnliche Tumoren als AFX oder PDS diagnostiziert wurden. Wahrscheinlich ist ein Ursprung aus den Bindegewebszellen, diskutiert werden auch Plattenepithelzellen der Haut.
Der Begriff PDS wird für Tumoren verwendet, die mikroskopisch dem AFX entsprechen. Allerdings erfassen diese Tumoren Anteile des tiefer gelegenen Unterhautfettgewebes und weisen nekrotische (=abgestorben) Tumoranteile und/oder eine Ausbreitung entlang von Nerven oder in Gefäßen auf. Als Risikofaktoren sind UV-Licht, radioaktive Strahlung, eine Immunsuppression (=Unterdrückung des körpereigenen Abwehrsystems) und genetische Erkrankungen (z.B. vererbte Defekte der DNA-Reparaturmechanismen, die verhindern, dass Schäden der Haut durch Sonneneinstrahlung regenerieren können) beschrieben.
Das atypische Fibroxanthom (AFX) und das pleomorphe dermale Sarkom (PDS) sind seltene bösartige Tumoren der Haut.
Sie unterscheiden sich im Wesentlichen durch ihre Tiefenausdehnung und das Vorliegen von Tumornekrosen (=abgestorbene Tumoranteile), Nerven-oder Gefäßausbreitung beim PDS.
- REFERENZEN
- Koelsche C, Stichel D, Griewank KG, Schrimpf D, Reuss DE, Bewerunge-Hudler M, et al. Genome-wide methylation profiling and copy number analysis in atypical fibroxanthomas and pleomorphic dermal sarcomas indicate a similar molecular phenotype. Clin Sarcoma Res. 2019;9:2.
- Helwig EB, May D. Atypical fibroxanthoma of the skin with metastasis. Cancer. 1986;57(2):368-76.
- Mentzel T, Requena L, Brenn T. Atypical Fibroxanthoma Revisited. Surg Pathol Clin. 2017;10(2):319-35.
- Dei Tos AP, Maestro R, Doglioni C, Gasparotto D, Boiocchi M, Laurino L, et al. Ultraviolet-induced p53 mutations in atypical fibroxanthoma. Am J Pathol. 1994;145(1):11-7.
- Persa OD, Loquai C, Wobser M, Baltaci M, Dengler S, Kreuter A, et al. Extended surgical safety margins and ulceration are associated with an improved prognosis in pleomorphic dermal sarcomas. J Eur Acad Dermatol Venereol. 2019.
- Bitel A, Schonlebe J, Kronert C, Wollina U. Atypical fibroxanthoma: An analysis of 105 tumors. Dermatol Ther. 2020:e13962.
- Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma: adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am J Surg Pathol. 2012;36(9):1317-26.
- Tolkachjov SN, Kelley BF, Alahdab F, Erwin PJ, Brewer JD. Atypical fibroxanthoma: Systematic review and meta-analysis of treatment with Mohs micrographic surgery or excision. J Am Acad Dermatol. 2018;79(5):929-34 e6.
- Tardio JC, Pinedo F, Aramburu JA, Suarez-Massa D, Pampin A, Requena L, et al. Pleomorphic dermal sarcoma: a more aggressive neoplasm than previously estimated. J Cutan Pathol. 2016;43(2):101-12.
- Klein S, Persa OD, Mauch C, Noh KW, Pappesch R, Wagener-Ryczek S, et al. First report on two cases of pleomorphic dermal sarcoma successfully treated with immune checkpoint inhibitors. Oncoimmunology. 2019;8(12):e1665977.
- Klein S, Mauch C, Wagener-Ryczek S, Schoemmel M, Buettner R, Quaas A, et al. Immune-phenotyping of pleomorphic dermal sarcomas suggests this entity as a potential candidate for immunotherapy. Cancer Immunol Immunother. 2019.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Symptome von Sarkomen und seltenen Tumoren der Haut
Zuletzt aktualisiert: 14.01.2026 | Autor(in): Doris Helbig, Mirjana Ziemer
AFX und PDS treten typischerweise an Körperstellen auf, die über längere Zeit dem Licht ausgesetzt sind. Hierzu gehören am häufigsten der behaarte Kopf und das Gesicht. Es sind meist haut- bis fleischfarbene feste Knoten, die bis zu mehreren Zentimetern groß und ulzeriert (=geschwürig) sein können. Das Tumorwachstum variiert zwischen wenigen Wochen bis mehreren Monaten, wobei PDS oftmals über einen längeren Zeitraum entstehen. Der Altersgipfel bei Erstdiagnose liegt bei Patienten im 7. bis 8. Lebensjahrzehnt. Es sind überwiegend Männer betroffen. Bei Patienten mit Immunsuppression (=Unterdrückung des körpereigenen Abwehrsystems) oder speziellen genetischen Erkrankungen ist ein Auftreten bereits in früherem Alter möglich.
Die Prognose von AFX und PDS ist von der Eindringtiefe, der Beteiligung tiefliegender Strukturen sowie der Ausbreitung entlang von Nerven oder in Gefäße abhängig.
Bei AFX kann nach vollständiger chirurgischer Entfernung von einer Heilung ausgegangen werden. Das ist insbesondere dann der Fall, wenn mittels sogenannter mikroskopisch kontrollierter Chirurgie (MKC) operiert wurde. Bei der MKC werden die Schnittränder unter dem Mikroskop lückenlos geprüft. Sollten zu einer Seite Tumorausläufer in den Schnittrand reichen, kann am nächsten Tag nach der Operation an dieser Stelle nachgeschnitten werden. So kann gewebesparend operiert werden und ein Rückfall weitgehend verhindert werden. Bei PDS hingegen sind Rückfälle (=Rezidive) beschrieben, wobei diese in der Regel innerhalb der ersten beiden Jahre nach Erstoperation auftreten. Bei Entfernung mit einem Sicherheitsabstand von 2 cm ist das Rückfallrisiko auch bei PDS gering. Eine Metastasierung ist beim PDS möglich. Die Metastasierung erfolgt vor allem in die Haut und die Lymphknoten der Tumorregion, seltener kommt es zu Fernmetastasen, zum Beispiel in der Lunge. Patienten mit begleitenden Blutkrebserkrankungen scheinen dabei häufiger betroffen zu sein.
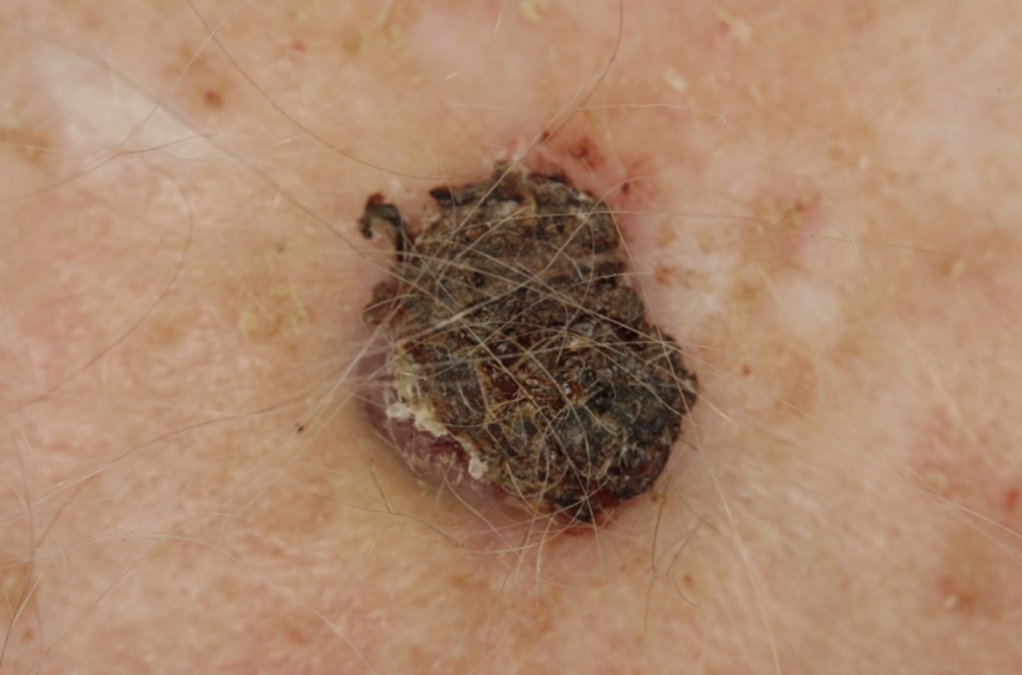
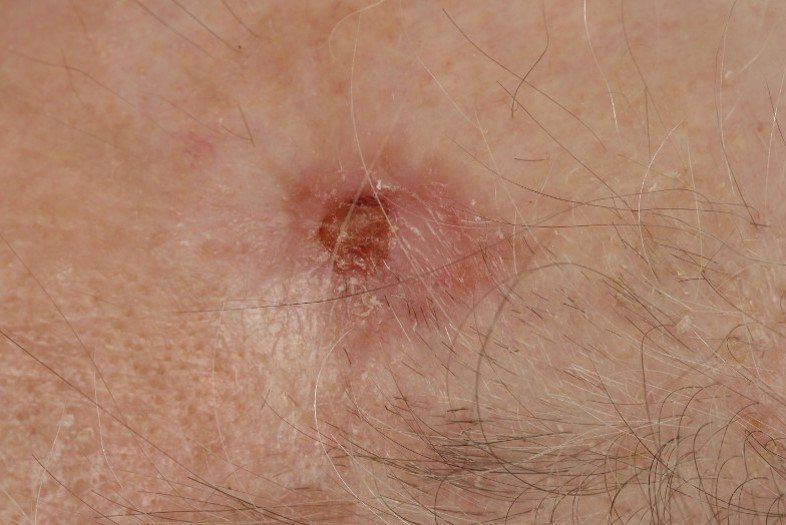
- REFERENZEN
- Koelsche C, Stichel D, Griewank KG, Schrimpf D, Reuss DE, Bewerunge-Hudler M, et al. Genome-wide methylation profiling and copy number analysis in atypical fibroxanthomas and pleomorphic dermal sarcomas indicate a similar molecular phenotype. Clin Sarcoma Res. 2019;9:2.
- Helwig EB, May D. Atypical fibroxanthoma of the skin with metastasis. Cancer. 1986;57(2):368-76.
- Mentzel T, Requena L, Brenn T. Atypical Fibroxanthoma Revisited. Surg Pathol Clin. 2017;10(2):319-35.
- Dei Tos AP, Maestro R, Doglioni C, Gasparotto D, Boiocchi M, Laurino L, et al. Ultraviolet-induced p53 mutations in atypical fibroxanthoma. Am J Pathol. 1994;145(1):11-7.
- Persa OD, Loquai C, Wobser M, Baltaci M, Dengler S, Kreuter A, et al. Extended surgical safety margins and ulceration are associated with an improved prognosis in pleomorphic dermal sarcomas. J Eur Acad Dermatol Venereol. 2019.
- Bitel A, Schonlebe J, Kronert C, Wollina U. Atypical fibroxanthoma: An analysis of 105 tumors. Dermatol Ther. 2020:e13962.
- Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma: adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am J Surg Pathol. 2012;36(9):1317-26.
- Tolkachjov SN, Kelley BF, Alahdab F, Erwin PJ, Brewer JD. Atypical fibroxanthoma: Systematic review and meta-analysis of treatment with Mohs micrographic surgery or excision. J Am Acad Dermatol. 2018;79(5):929-34 e6.
- Tardio JC, Pinedo F, Aramburu JA, Suarez-Massa D, Pampin A, Requena L, et al. Pleomorphic dermal sarcoma: a more aggressive neoplasm than previously estimated. J Cutan Pathol. 2016;43(2):101-12.
- Klein S, Persa OD, Mauch C, Noh KW, Pappesch R, Wagener-Ryczek S, et al. First report on two cases of pleomorphic dermal sarcoma successfully treated with immune checkpoint inhibitors. Oncoimmunology. 2019;8(12):e1665977.
- Klein S, Mauch C, Wagener-Ryczek S, Schoemmel M, Buettner R, Quaas A, et al. Immune-phenotyping of pleomorphic dermal sarcomas suggests this entity as a potential candidate for immunotherapy. Cancer Immunol Immunother. 2019.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Behandlung von Sarkomen und seltenen Tumoren der Haut
Zuletzt aktualisiert: 14.01.2026 | Autor(in): Doris Helbig, Mirjana Ziemer
Therapie
Für das AFX und das PDS soll unter heilender Absicht (=kurativer Intention) eine komplette operative Entfernung mit anschließender feingeweblicher (mikroskopischer) Untersuchung erfolgen. Wenn möglich soll eine sogenannte mikrographisch kontrollierte Exzision (MKC) erfolgen, sowie ein entsprechender Sicherheitsabstand zum Tumorrand eingehalten werden.
Beim AFX ist eine mikrographisch (also per Mikroskopie) kontrollierte Resektion knapp im Gesunden ausreichend. Im Falle des PDS sollte der Sicherheitsabstand auf bis zu 2 cm erweitert werden, um das Risiko eines erneuten lokalen Wachstums des Tumors (=Rückfallrisiko) zu senken. Dabei müssen anatomische, funktionelle und ästhetische Aspekte vor allem im Gesicht berücksichtigt werden, da bei Operationen mit großen Sicherheitsabständen Einbußen in der Funktion der Körperregion (z.B. Einschränkung der Mimik, Mundöffnung oder Lidschluss) oder der ästhetischen Gesichtsform (Veränderung der Gesichtsform, Asymmetrie der Gesichtshälften, veränderte Gesichtsproportionen) drohen. Die endgültige Entscheidung für abweichende Sicherheitsabstände sollte der Operateur im Einverständnis mit dem informierten Patienten treffen, auch in Abhängigkeit der speziellen Lage des Tumors.
PDS sind seltene Hauttumoren und daher wird empfohlen, diese im interdisziplinären Hauttumorboard zu besprechen, unabhängig vom Stadium. Bei Patienten mit dieser seltenen Erkrankung sollte die Behandlung in einem zertifizierten Hautkrebszentrum durchgeführt werden. Informationen zu einem zertifizierten Hautkrebszentrum in Ihrer Nähe finden Sie unter https://www.oncomap.de/.
Es gibt keine publizierten Daten zur Empfindlichkeit von AFX/PDS auf eine Bestrahlungsbehandlung. Ist jedoch eine vollständige Operation des Tumors nicht möglich, kann eine Nachbestrahlung des Resttumorareals erwogen werden. Eventuell besteht auch ein Nutzen einer Nachbestrahlung bei vollständig operierten PDS zur Vorbeugung eines erneuten Tumorwachstums
Therapie mit Medikamenten
Eine wirksame Standardtherapie mit Medikamenten fortgeschrittener AFX/PDS ist nicht bekannt. Therapieempfehlungen für nicht operierbare oder metastasierte Patienten sollten im Rahmen eines interdisziplinären Tumorboards individuell besprochen werden. In die Entscheidung könnten neben den klinischen Aspekten (=Beschwerden, erkennbare Symptome), Ergebnisse molekulargenetischer Untersuchungen und mikroskopische Tumoreigenschaften einfließen.
Es gibt vereinzelte Berichte über Behandlungsversuche mit verschiedenen Chemotherapien. Auch kann eine immunmodulierende Therapie mit einem sogenannten Checkpointinhibitor wie z.B. einem anti-PD-1 Antikörper erwogen werden. Jedoch sind diese Therapien nicht für das AFX / PDS zugelassen und erfordern deshalb eine vorherige Klärung der Kostenübernahme durch die Krankenkasse.
Die Therapie der ersten Wahl ist die vollständige, jedoch möglichst funktionserhaltende chirurgische Entfernung.
Eine wirksame Standardtherapie mit Medikamenten fortgeschrittener AFX/PDS ist nicht bekannt.
Der Einsatz von Checkpointinhibitoren könnte vielversprechend sein, ist jedoch nicht zugelassen.
- REFERENZEN
- Koelsche C, Stichel D, Griewank KG, Schrimpf D, Reuss DE, Bewerunge-Hudler M, et al. Genome-wide methylation profiling and copy number analysis in atypical fibroxanthomas and pleomorphic dermal sarcomas indicate a similar molecular phenotype. Clin Sarcoma Res. 2019;9:2.
- Helwig EB, May D. Atypical fibroxanthoma of the skin with metastasis. Cancer. 1986;57(2):368-76.
- Mentzel T, Requena L, Brenn T. Atypical Fibroxanthoma Revisited. Surg Pathol Clin. 2017;10(2):319-35.
- Dei Tos AP, Maestro R, Doglioni C, Gasparotto D, Boiocchi M, Laurino L, et al. Ultraviolet-induced p53 mutations in atypical fibroxanthoma. Am J Pathol. 1994;145(1):11-7.
- Persa OD, Loquai C, Wobser M, Baltaci M, Dengler S, Kreuter A, et al. Extended surgical safety margins and ulceration are associated with an improved prognosis in pleomorphic dermal sarcomas. J Eur Acad Dermatol Venereol. 2019.
- Bitel A, Schonlebe J, Kronert C, Wollina U. Atypical fibroxanthoma: An analysis of 105 tumors. Dermatol Ther. 2020:e13962.
- Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma: adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am J Surg Pathol. 2012;36(9):1317-26.
- Tolkachjov SN, Kelley BF, Alahdab F, Erwin PJ, Brewer JD. Atypical fibroxanthoma: Systematic review and meta-analysis of treatment with Mohs micrographic surgery or excision. J Am Acad Dermatol. 2018;79(5):929-34 e6.
- Tardio JC, Pinedo F, Aramburu JA, Suarez-Massa D, Pampin A, Requena L, et al. Pleomorphic dermal sarcoma: a more aggressive neoplasm than previously estimated. J Cutan Pathol. 2016;43(2):101-12.
- Klein S, Persa OD, Mauch C, Noh KW, Pappesch R, Wagener-Ryczek S, et al. First report on two cases of pleomorphic dermal sarcoma successfully treated with immune checkpoint inhibitors. Oncoimmunology. 2019;8(12):e1665977.
- Klein S, Mauch C, Wagener-Ryczek S, Schoemmel M, Buettner R, Quaas A, et al. Immune-phenotyping of pleomorphic dermal sarcomas suggests this entity as a potential candidate for immunotherapy. Cancer Immunol Immunother. 2019.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Nachsorge bei Sarkomen und seltenen Tumoren der Haut
Zuletzt aktualisiert: 14.01.2026 | Autor(in): Doris Helbig, Mirjana Ziemer
Die Nachsorge von Patienten mit AFX und PDS mit einer frühzeitigen Entdeckung von Rückfällen ist wichtig. Bei Patienten mit PDS ist außerdem die frühzeitige Erfassung von Lymphknoten- oder Fernmetastasen von Bedeutung.
Bei AFX werden halbjährliche klinische Untersuchungen einschließlich der Abtastung der Lymphknoten der Region, innerhalb der ersten beiden Jahre empfohlen. Anschließend jährlich für mindestens fünf Jahre (Tabelle 1).
Bei PDS werden in Abständen von 3 Monaten klinische Untersuchungen einschließlich der Abtastung der Lymphknoten der Region innerhalb der ersten beiden Jahre empfohlen. Im Anschluss werden halbjährliche Abstände für mindestens fünf Jahre empfohlen.
Bei PDS sollte in 6-monatlichen Abständen eine Ultraschalluntersuchung der ursprünglichen Tumorregion sowie der lokalen Lymphknoten erfolgen. Weiterführende Untersuchungen wie Computertomografie (CT) oder Magnetresonanztomografie (MRT) sind speziellen Situationen und Fragestellungen (so z.B. bei auffälligem Befund im Ultraschall) vorbehalten.
Nachsorgeschema für das atypische Fibroxanthom (AFX) und das pleomorphe dermale Sarkom (PDS)
| Jahr 1 & 2 | Jahr 3 – 8 (5 Jahre) | |
|---|---|---|
| AFX | Alle 6 Monate | Jährlich |
| PDS | Alle 3 Monate | Alle 6 Monate |
AFX und PDS treten am häufigsten am behaarten Kopf und im Gesicht auf.
Der Altersgipfel bei Erstdiagnose liegt im 7. bis 8. Lebensjahrzehnt, wobei überwiegend Männer betroffen sind.
Bei AFX kann nach vollständiger chirurgischer Entfernung von einer Heilung ausgegangen werden. Rückfälle treten zumeist in den ersten 2 Jahren auf und sind häufiger bei PDS.
- REFERENZEN
- Koelsche C, Stichel D, Griewank KG, Schrimpf D, Reuss DE, Bewerunge-Hudler M, et al. Genome-wide methylation profiling and copy number analysis in atypical fibroxanthomas and pleomorphic dermal sarcomas indicate a similar molecular phenotype. Clin Sarcoma Res. 2019;9:2.
- Helwig EB, May D. Atypical fibroxanthoma of the skin with metastasis. Cancer. 1986;57(2):368-76.
- Mentzel T, Requena L, Brenn T. Atypical Fibroxanthoma Revisited. Surg Pathol Clin. 2017;10(2):319-35.
- Dei Tos AP, Maestro R, Doglioni C, Gasparotto D, Boiocchi M, Laurino L, et al. Ultraviolet-induced p53 mutations in atypical fibroxanthoma. Am J Pathol. 1994;145(1):11-7.
- Persa OD, Loquai C, Wobser M, Baltaci M, Dengler S, Kreuter A, et al. Extended surgical safety margins and ulceration are associated with an improved prognosis in pleomorphic dermal sarcomas. J Eur Acad Dermatol Venereol. 2019.
- Bitel A, Schonlebe J, Kronert C, Wollina U. Atypical fibroxanthoma: An analysis of 105 tumors. Dermatol Ther. 2020:e13962.
- Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma: adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am J Surg Pathol. 2012;36(9):1317-26.
- Tolkachjov SN, Kelley BF, Alahdab F, Erwin PJ, Brewer JD. Atypical fibroxanthoma: Systematic review and meta-analysis of treatment with Mohs micrographic surgery or excision. J Am Acad Dermatol. 2018;79(5):929-34 e6.
- Tardio JC, Pinedo F, Aramburu JA, Suarez-Massa D, Pampin A, Requena L, et al. Pleomorphic dermal sarcoma: a more aggressive neoplasm than previously estimated. J Cutan Pathol. 2016;43(2):101-12.
- Klein S, Persa OD, Mauch C, Noh KW, Pappesch R, Wagener-Ryczek S, et al. First report on two cases of pleomorphic dermal sarcoma successfully treated with immune checkpoint inhibitors. Oncoimmunology. 2019;8(12):e1665977.
- Klein S, Mauch C, Wagener-Ryczek S, Schoemmel M, Buettner R, Quaas A, et al. Immune-phenotyping of pleomorphic dermal sarcomas suggests this entity as a potential candidate for immunotherapy. Cancer Immunol Immunother. 2019.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Kaposi-Sarkom
Zuletzt aktualisiert: 10.04.2026 | Autor(in): Prof. Dr. med. Stefan Esser
Definition, Ursachen und Einteilung in unterschiedliche Subtypen
Das Kaposi-Sarkom (KS) ist eine seltene, maligne, von lymphatischen Endothelzellen ausgehende, an mehreren Orten auftretende Gefäßerkrankung.
Sie befällt vor allem Haut und Schleimhäute, aber auch das lymphatische System und innere Organe wie den Gastrointestinaltrakt, die Lunge oder die Leber. 1981 wurde erstmals eine neue, aggressiv verlaufende Variante bei jungen Menschen mit HIV-Infektion als AIDS-Erkrankung beschrieben [1]. KS werden durch das humane Herpes-virus 8 (HHV-8) verursacht. Daher wird es auch als Kaposi-Sarkom-assoziiertes Herpesvirus (KSHV) bezeichnet [2]. Eine generelle Immunschwäche begünstigt die Entwicklung von KS.
Es gibt fünf epidemiologische Unterformen des KS. Diese treten in verschiedenen Patientengruppen vermehrt auf. Sie unterscheiden sich im klinischen Verlauf und ihrer Prognose:
- Klassisches (sporadisches) Kaposi-Sarkom
Das klassische KS tritt überwiegend bei älteren (>60 Jahre) Männern (Mann:Frau Ratio 2:1 bis 17:1) osteuropäischer und mediterraner Herkunft auf. Erstmalig tritt es meist im Lebensalter von über 60 Jahren auf. Die Risikofaktoren für die Entstehung eines KS vom klassischen Subtyp umfassen vorrangig eine vorangegangene HHV-8-Infektion sowie erhöhtes Lebensalter. Die Sterberate ist bei Patienten mit klassischem KS im Vergleich zur Normalbevölkerung nach bisherigem Kenntnisstand nicht wesentlich erhöht [4].
- Afrikanisches, endemisches Kaposi-Sarkom
Das endemische, nicht mit einer HIV-Infektion assoziierte KS ist in Äquatorialafrika vor allem auch unter Kindern und adoleszenten Männern verbreitet. Anhand von Ausbreitungsmuster und Aggressivität werden vier klinische Verlaufsformen unterschieden:
- Benigne Verlaufsform: Langsam und wenig aggressiv fortschreitende noduläre KS-Läsionen der Haut, ähnlich denen beim klassischen KS; vorwiegend bei Männern mittleren Alters.
- Lokal aggressive Verlaufsform: Kutane KS-Läsionen mit aggressiver Infiltration in Weichgewebe und Knochen. Meist tödlicher Ausgang innerhalb von fünf bis sieben Jahren.
- Diffus aggressive Verlaufsform: Diffuser mukokutaner (=betrifft die Schleimhäute) und viszeraler Befall (die Eingeweide betreffend); ungünstige Prognose.
- Fulminante Verlaufsform: Vorrangig Lymphadenopathie (=tastbare Vergrößerung eines Lymphknotens) und Befall der inneren Organe, selten Hautbeteiligung; fulminant aggressiver Krankheitsverlauf; vorrangig bei Kleinkindern.
Das endemische KS lag in Zentralafrika bei 9% aller landesweiten Krebserkrankungen. Mit dem Ausbruch der HIV-Pandemie stieg allerdings die Inzidenz des HIV-assoziierten KS vor allem in Subsahara-Afrika drastisch [5].
- Kaposi-Sarkom bei iatrogener (=ärztlich verursachter) Immunsuppression
Unter langfristiger Immunsuppression können sich iatrogene KS entwickeln, die sich nach Absetzen, Reduktion oder Umsetzen der immunsuppressiven Therapie wieder zurückbilden können. Bei mehr als 5% der organtransplantierten Patienten, die nachfolgend ein Malignom entwickelten, trat ein KS auf [16]. Das Risiko ein KS zu entwickeln ist bei Organtransplantierten im Vergleich zur Normalbevölkerung um das 50- bis 500-fache erhöht [6].
- HIV-assoziiertes, epidemisches Kaposi-Sarkom
Das epidemische, HIV-assoziierte KS ist die häufigste AIDS-definierende bösartige Neubildung (Neoplasie). Seit Beginn der HIV-Pandemie ist sie auch der häufigste klinische Subtyp des KS. Der Verlauf ist äußerst variabel und reicht von einzelnen über Jahre stationären Läsionen bis hin zu tödlichen Verläufen [7]. Besonders nicht oder nicht ausreichend antiretroviral behandelte HIV-Infizierte in fortgeschrittenen Stadien mit schwerer Immundefizienz erkranken am HIV-assoziierten KS. Nach Einleitung einer antiretroviralen Therapie (ART) bilden sich HIV-assoziierte KS häufig ohne zusätzliche Behandlung zurück. Im Zusammenhang mit dem Immun-Rekonstitutions-Inflammations-Syndrom (IRIS) [8] wurden unerwartet schwere Verläufe und/oder neu auftretende KS beschrieben.
Trotz breitem Einsatz effektiver ART treten auch neue KS oder KS-Rezidive bei gutem Immunstatus auf. Das Risiko ein KS zu entwickeln ist für Menschen mit HIV-Infektion 30-80-mal höher als in der Allgemeinbevölkerung. Möglicherweise entstehen bei älteren HIV-Patienten neue KS, weil ihr Immunsystem im Alter schwächer wird. Die Sterblichkeit des HIV-assoziierten KS steht in Zusammenhang mit der CD4-Zellzahl sowie mit der konsequenten Einnahme der ART [7,9].
- Kaposi-Sarkom bei Männern, die Sex mit Männern haben, ohne HIV-Infektion
Seit einigen Jahren wird zunehmend über KS bei jüngeren HIV-negativen Männern, die Sex mit Männern (MSM), aus geographischen Regionen mit einer niedrigen HHV-8-Seroprävalenz (z.B. Frankreich, England oder Deutschland) berichtet. Der Verlauf ist ähnlich wie beim klassischen KS eher schmerzunempfindlich, die Hautläsionen kommen an der gesamten Körperoberfläche vor. Ein viszeraler Befall oder Organbefall ist sehr selten. Aufgrund dieser Besonderheiten und Unterschieden zu den bisher 4 anerkannten Subtypen des KS wird diese Form neu als zusätzlicher (fünfter) epidemiologischer Subtyp klassifiziert [10].
Klinik, Diagnosestellung und Ausbreitungsdiagnostik
Anfänglich entwickeln sich bei allen Formen asymptomatische, lividrote Flecken oder Knoten. Einzelne Tumoren können über Jahre unverändert bleiben oder sich in wenigen Wochen rasch ausbreiten und an Zahl und Größe zunehmen. Ausgeprägte Schwellungen ganzer Extremitäten, des Skrotums oder des Gesichts aufgrund einer Lymphabflussstörung kommen vor. Typisch sind Einblutungen in die Tumorumgebung, die als gelb-grüne Verfärbungen auffallen. Weit fortgeschrittene Einzeltumoren können zentral aufbrechen und bluten. Auch stark verhornte (warzenförmige) Formen, die den Gefäßcharakter der Tumoren völlig verbergen, treten betont an den unteren Extremitäten auf. An der Mundschleimhaut ist besonders der harte Gaumen betroffen. Auch innere Organe, besonders der Verdauungstrakt und die Lunge können befallen werden.
Lokale und systemische Therapie
Ziel der Behandlung ist in der Regel die Rückbildung der Läsionen, die Kontrolle über den Krankheitsverlauf und die Reduktion der Symptome mit Erhalt der Lebensqualität und der Lebenserwartung.
Bisher gibt es kein allgemein anerkanntes „Standardtherapieschema" zur Behandlung des KS.
Sowohl lokale als auch systemische Behandlungsmethoden mit unterschiedlichen Wirkmechanismen stehen für die Therapie von KS zur Verfügung und können auch kombiniert eingesetzt werden.
Nicht immer ist eine sofortige spezifische Behandlung nach gesicherter Diagnose bei guter Prognose und fehlendem Leidensdruck des Patienten bei entsprechender Nachbeobachtung erforderlich.
Bei rasch fortschreitendem oder aggressivem Verlauf, dem Auftreten von Symptomen und/oder funktionellen Beeinträchtigungen soll unabhängig vom KS-Typ sofort eine gezielte Behandlung eingeleitet werden. Bei bisher noch nicht oder nicht effektiv antiretroviral behandelter HIV-Infektion soll umgehend eine ART eingeleitet oder optimiert werden.
Das Kaposi-Sarkom (KS) ist eine seltene, maligne Lymphgefäßerkrankung mit fünf Unterformen, die in spezifischen Patientengruppen vermehrt auftreten. Sie unterscheiden sich im klinischen Verlauf und ihrer Prognose.
Bei rasch fortschreitendem oder aggressivem Verlauf, dem Auftreten von Symptomen und/oder funktionellen Beeinträchtigungen soll unabhängig vom KS-Typ sofort eine gezielte Behandlung eingeleitet werden.
KS werden durch Humane Herpes-Virus-Infektionen (HHV8) verursacht.
Immunsuppression und Immundefizienz begünstigen die Entwicklung von KS.
- REFERENZEN
- 1. Friedman-Kien AE Disseminated Kaposi's sarcoma syndrome in young homosexual men. J Am Acad Dermatol 1981; 5: 468-471.
- 2. Cesarman E, Damania B, Krown SE, Martin J et al. Kaposi sarcoma. Nat Rev Dis Primers 2019; 5(1): 9.
- 3. Dedicoat M, Newton R. Review of the distribution of Kaposi's sarcoma-associated herpesvirus (HHV-8) in Africa in relation to the incidence of Kaposi's sarcoma. Br J Cancer 2003; 88(1): 1-3.
- 4. Franceschi S, Arniani S, Balzi D et al. Survival of classic Kaposi's sarcoma and risk of second cancer. Br J Cancer. 1996; 74: 1812-4.
- 5. Liu Z, Fang Q, Zuo J et al. The world-wide incidence of Kaposi's sarcoma in the HIV/AIDS era. HIV Med 2018; 19(5): 355-364.
- 6. Lebbe C, Legendre C, Frances C. Kaposi sarcoma in transplantation. Transplant Rev (Orlando). 2008; 22: 252-61.
- 7. Hoffmann C, Sabranski M, Esser S. HIV-Associated Kaposi's Sarcoma. Oncol Res Treat 2017; 40(3): 94-98.
- 8. Müller M, Wandel S, Colebunders R et al.; IeDEA Southern and Central Africa. Immune reconstitution inflammatory syndrome in patients starting antiretroviral therapy for HIV infection: a systematic review and meta-analysis. Lancet Infect Dis 2010; 10(4): 251-61.
- 9. Palich R, Veyri M, Valantin MA, et al.; CancerVIH Study Group. Recurrence and Occurrence of Kaposi's Sarcoma in Patients Living With Human Immunodeficiency Virus (HIV) and on Antiretroviral Therapy, Despite Suppressed HIV Viremia. Clin Infect Dis 2020; 70(11): 2435-2438.
- 10. Denis D, Seta V, Regnier-Rosencher E et al. A fifth subtype of Kaposi's sarcoma, classic Kaposi's sarcoma in men who have sex with men: a cohort study in Paris. J Eur Acad Dermatol Venereol 2018; 32(8): 1377-1384.
- 11. Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S1-Leitlinie Kaposi-Sarkom (KS)
- 12. Di Trolio R, Di Lorenzo G, Delfino M et al. Role of pegylated lyposomal doxorubicin (PLD) in systemic Kaposi´s sarcoma: a systematic review. Int J Immunopathol Pharmacol 2006; 19: 253-63.
- 13. Kreuter A, Rasokat H, Klouche M, et al. Liposomal pegylated doxorubicin versus low-dose recombinant interferon Alfa-2a in the treatment of advanced classic Kaposi's sarcoma; retrospective analysis of three German centers. Cancer Invest. 2005; 23(8): 653-9.
- 14. Uldrick TS, Gonçalves PH, Abdul-Hay M, et al. Assessment of the Safety of Pembrolizumab in Patients With HIV and Advanced Cancer-A Phase 1 Study. JAMA Oncol 2019; 5(9): 1332-9.
INTERESSENSKONFLIKTE
Der Autor/die Autorin hat keine Interessenskonflikte angegeben.
Link copied to clipboard!